Source:
http://www.cmaj.ca/content/171/3/251/F2.large.jpg
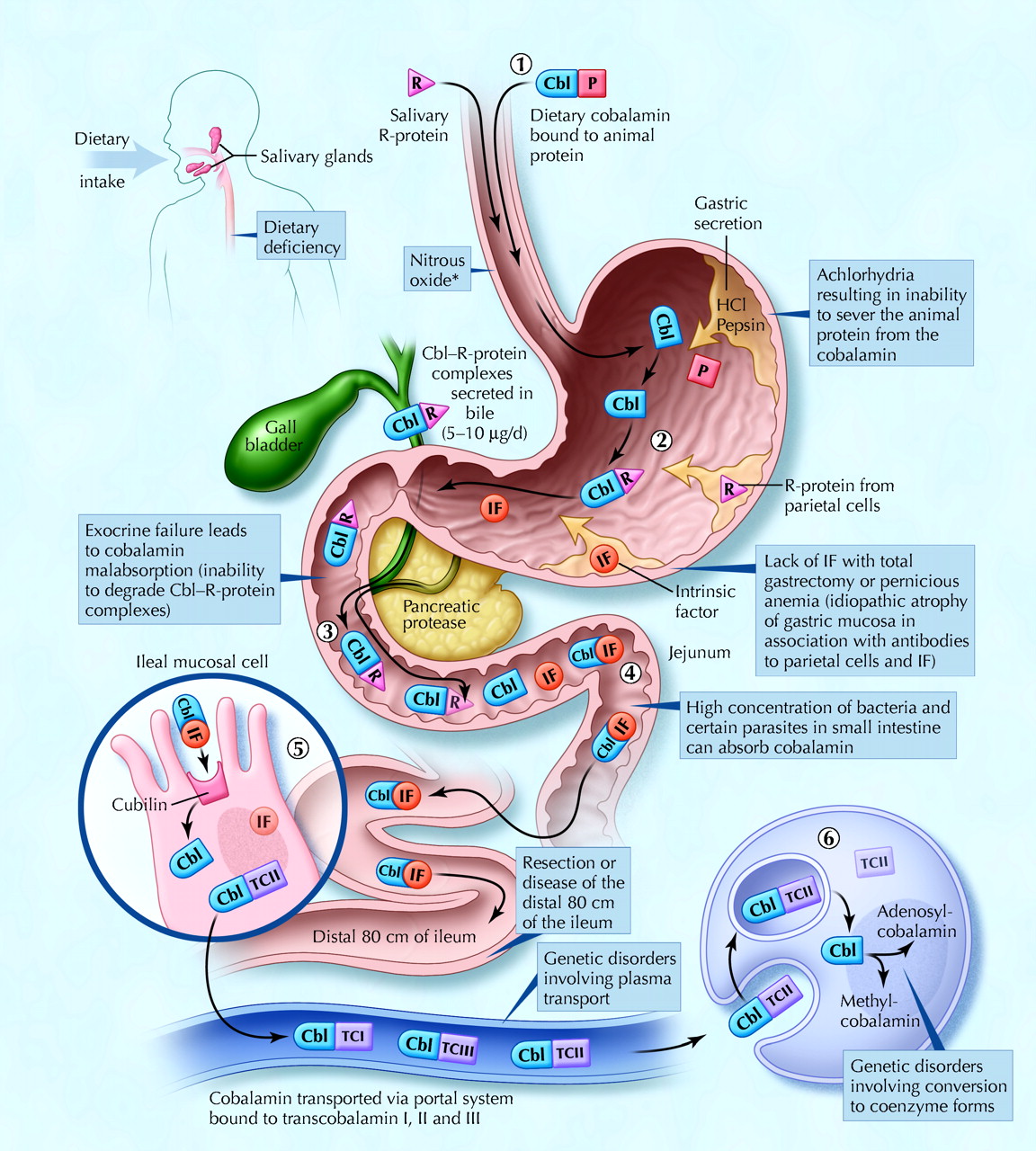
Fig. 1: Cobalamin metabolism and corresponding causes of deficiency. Causes of cobalamin deficiency are shown in blue. The metabolic pathway starts when (1) dietary cobalamin (Cbl), obtained through animal foods, enters the stomach bound to animal proteins (P). (2) Pepsin and hydrochloric acid (HCl) in the stomach sever the animal protein, releasing free cobalamin. Most of the free cobalamin is then bound to R-protein (R), which is released from the parietal and salivary cells. Intrinsic factor (IF) is also secreted in the stomach, but its binding to cobalamin is weak in the presence of gastric and salivary R-protein. (3) In the duodenum, dietary cobalamin bound to R-protein is joined by cobalamin–R-protein complexes that have been secreted in the bile. Pancreatic enzymes degrade both biliary and dietary cobalamin–R-protein complexes, releasing free cobalamin. (4) The cobalamin then binds with intrinsic factor. The cobalamin–intrinsic factor complex remains undisturbed until the distal 80 cm of the ileum, where (5) it attaches to mucosal cell receptors (cubilin) and the cobalamin is bound to transport proteins known as transcobalamin I, II and III (TCI, TCII and TCIII). Transcobalamin II, although it represents only a small fraction (about 10%) of the transcobalamins, is the most important because it is able to deliver cobalamin to all cells in the body. The cobalamin is subsequently transported systemically via the portal system. (6) Within each cell, the transcobalamin II–cobalamin complex is taken up by means of endocytosis and the cobalamin is liberated and then converted enzymatically into its 2 coenzyme forms, methylcobalamin and adenosylcobalamin (this process is shown in greater detail in Fig. 2).
*Nitrous oxide, a general anesthetic, causes multiple defects in cobalamin use, most of which are intracellular and clinically relevant only in people who have low or borderline-low serum cobalamin levels. Photo: Christine Kenney
Source:
http://www.cmaj.ca/content/171/3/251/F3.large.jpg
Cellular uptake and processing of cobalamin. Cobalamin (Cbl) bound to the transport protein transcobalamin II (TCII) enters cells by means of transcobalamin II receptor-mediated endocytosis. Lysosomal enzymes degrade the transcobalamin II, thereby freeing the cobalamin. Cob(III)alamin (CBL
III) represents the most oxidized form of cobalamin, and cob(II)alamin (CBL
II) and cob(I)alamin (CBL
I) represent reduced forms. In the mitochondria, cobalamin is converted to adenosylcobalamin (AdoCbl), a coenzyme involved in the conversion of methylmalonyl-CoA (MM-CoA) to succinyl-CoA. In the cytoplasm, cobalamin functions as a coenzyme for the reaction catalyzed by methionine synthase. PteGlu = folic acid, MeCbl = methylcobalamin.
B. Intracytoplasmic biochemical pathways involving cobalamin. BHMT = betaine-homocysteine S-methyltransferase, NADP = nicotinamide adenine dinucleotide phosphate, NADPH = reduced form of NADP. Photo: Christine Kenney
Reference:
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4219318/
Intracellular metabolism of cobalamin and intersecting pathways. The
steps of the intracellular cobalamin metabolism, their complementation
groups and corresponding genes are shown. The folate cycle, 1-carbon
metabolism and the betaine-homocysteine methyltransferase reaction are
shown for their importance in the pathophysiology and treatment of cblC
disease.
Abbreviations.
AdoCbl: 5’-deoxyadenosylcobalamin;
Cbl: Cobalamin;
Cbl lII: Dietary cobalamin contains a cobalt in the 3+ state (oxidized) and it is reduced to
Cbl II and
Cbl I, for its use in coenzyme synthesis.
CBS: Cystathionine beta synthase;
Gly: Glycine;
LMBRD1: Lysosomal cobalamin exporter, defective in methylmalonic acidemia and homocystinuria, cblF type;
MeCbl: Methylcobalamin;
MMAA: Gene responsible for methylmalonic acidemia, cblA type;
MMAB: Gene responsible for methylmalonic acidemia, cblB type;
MMACHC: Gene responsible for methylmalonic acidemia and homocystinuria, cblC type;
MMADHC: Gene responsible for methylmalonic acidemia and/or homocystinuria, cblD type;
MUT: methylmalonyl-CoA mutase;
MTR: 5-methyltetrahydrofolate-homocysteine S-methyltransferase also known as methionine synthase;
MTRR: Methionine synthase reductase;
SAM:
S-adenosylmethionine;
Ser: Serine;
THF: Tetrahydrofolate
Source:
http://www.cmaj.ca/content/171/3/251/F4.large.jpg
Source:
http://www.cmaj.ca/content/171/3/251/F5.large.jpg
Diagnostic process for cobalamin deficiency. Photo: Christine Kenney
Reference:
Emmanuel Andrès,
Noureddine Henoun Loukili,
Esther Noel,
Georges Kaltenbach,
Maher Ben Abdelgheni,
Anne Elisabeth Perrin,
Marie Noblet-Dick,
Frédéric Maloisel,
Jean-Louis Schlienger,
Jean-Frédéric Blicklé. Vitamin B12 (cobalamin) deficiency in elderly patients. CMAJ August 3, 2004 vol. 171 no. 3 doi:
10.1503/cmaj.1031155
| Table 1. Conditions Reported to Improve by Coenzyme Forms of B12 |
|---|
| |
Therapeutic dose recommended by Kelly |
Conditions reported to improve from treatment,A |
| Methylcobalamin |
1500-6000 µg/day |
Diabetic neuropathy
Infertility
Hyperhomocysteinemia in diabetes
Sleep disordersB
Bell's Palsy |
Adenosylcobalamin, aka:
5'-deoxyadenosylcobalamin, dibencozide,
coenzyme B12, cobamamide, and cobinamide
|
1000-6000 µg/day |
Neurological problems secondary to anorexia
Hepatitis A
Viral hepatitisB |
Detailed mechanism and functions of methylcobalamin and adenosylcobalamin and their link with cytokines
http://emedicine.medscape.com/article/1152670-overview#a4
Background
The association of anemia and gastrointestinal and neurologic
abnormalities referable to the brain, spinal cord, and peripheral nerves
has been recognized in several clinical and postmortem case reports and
series by Combe, Addison, and Fenwick since the early 19th century. In
1877, Gardner and Osler coined the term
pernicious anemia (PA) to describe a patient with progressive arm numbness and difficulty with buttoning and using tools.
[1] Liechtenstein in 1884 reported the association of PA and spinal cord disease but attributed both to tabes dorsalis.
[2] Lichtheim in 1887
[3] and Minnich in 1892
[4] recognized the histologic differences in the spinal cord between PA and tabes dorsalis.
In 1900, Russell et al coined the term
subacute combined degeneration of the spinal cord.
[5] In 1926, Minot and Murphy fed PA patients a half-pound of calf liver daily, for which they received the Nobel Prize.
[6] In 1929, Castle distinguished the role of gastric (intrinsic) and dietary (extrinsic) factors in PA.
[7] In
1948, cyanocobalamin was isolated from the liver. The existence of
vitamin B-12 deficiency neuropathy was recognized in 1958. In 1955,
Lassen et al
[8] noted megaloblastic anemia secondary to prolonged nitrous oxide (N
2 O) exposure; the neurologic features were described in 1978 by Sahenk et al
[9] and Layzer et al.
[10]
See the image below.
 Vitamin B-12–associated neurological
diseases. Pernicious anemia. Characteristic lemon-yellow pallor with raw
beef tongue lacking filiform papillae. Photo from Forbes and Jackson
with permission.
Vitamin B-12–associated neurological
diseases. Pernicious anemia. Characteristic lemon-yellow pallor with raw
beef tongue lacking filiform papillae. Photo from Forbes and Jackson
with permission.
See
Hidden Clues to Diagnosing Nutritional Deficiencies, a Critical Images slideshow, to help identify clues to conditions associated with malnutrition.
Pathophysiology
Vitamin B-12 structure
Vitamin B-12 (cobalamin) is a complex molecule in which a
cobalt atom is contained in a corrin ring. Vitamin B-12 is available in
animal protein.
Body stores
Total body stores are 2-5 mg, of which half is stored in the
liver. The recommended daily intake is 2 mcg/d in adults; pregnant and
lactating women require 2.6 mcg/d. Children require 0.7 mcg/d and, in
adolescence, 2 mcg/d. Because vitamin B-12 is highly conserved through
the enterohepatic circulation, cobalamin deficiency from malabsorption
develops after 2-5 years and deficiency from dietary inadequacy in
vegetarians develops after 10-20 years. Its causes are mainly
nutritional and malabsorptive, PA being most common.
Physiology of absorption
After ingestion, the low stomach pH cleaves cobalamin from other dietary protein.
[11] The
free cobalamin binds to gastric R binder, a glycoprotein in saliva, and
the complex travels to the duodenum and jejunum, where pancreatic
peptidases digest the complex and release cobalamin. Free cobalamin can
then bind with gastric intrinsic factor (IF), a 50-kd glycoprotein
produced by the gastric parietal cells, the secretion of which parallels
that of hydrochloric acid. Hence, in states of achlorhydria, IF
secretion is reduced, leading to cobalamin deficiency. Importantly, only
99% of ingested cobalamin requires IF for absorption. Up to 1% of free
cobalamin is absorbed passively in the terminal ileum. This why oral
replacement with large vitamin B-12 doses is appropriate for PA.
Once bound with IF, vitamin B-12 is resistant to further
digestion. The complex travels to the distal ileum and binds to a
specific mucosal brush border receptor, cublin, which facilitates the
internalization of cobalamin-IF complex in an energy-dependent process.
Once internalized, IF is removed and cobalamin is transferred to other
transport proteins, transcobalamin I, II, and III (TCI, TCII, TCIII).
Eighty percent of cobalamin is bound to TCI/III, whose role in cobalamin
metabolism is unknown. The other 20% binds with TCII, the physiologic
transport protein produced by endothelial cells. Its half-life is 6-9
min, thus delivery to target tissues is rapid.
The cobalamin-TCII complex is secreted into the portal blood
where it is taken up mainly in the liver and bone marrow as well as
other tissues. Once in the cytoplasm, cobalamin is liberated from the
complex by lysosomal degradation. An enzyme-mediated reduction of the
cobalt occurs by cytoplasmic methylation to form methylcobalamin or by
mitochondrial adenosylation to form adenosylcobalamin, the 2
metabolically active forms of cobalamin.
Vitamin B-12 role in bone marrow function
In the cytoplasm, methylcobalamin (see image below) serves as
cofactor for methionine synthesis by allowing transfer of a methyl group
from 5-methyl-tetrahydrofolate (5-methyl-THF) to homocysteine (HC),
forming methionine and demethylated tetrahydrofolate (THF). This results
in reduction in serum homocysteine, which appears to be toxic to
endothelial cells. Methionine is further metabolized to
S-adenosylmethionine (SAM).
 Vitamin B-12–associated neurological
diseases. Cobalamin and folate metabolism. TS = thymidylate synthase,
DHFR = dihydrofolate reductase, SHMT = serine methyl-transferase.
Vitamin B-12–associated neurological
diseases. Cobalamin and folate metabolism. TS = thymidylate synthase,
DHFR = dihydrofolate reductase, SHMT = serine methyl-transferase.
THF is used for DNA synthesis. After conversion to its
polyglutamate form, THF participates in purine synthesis and the
conversion of deoxyuridylate (dUTP) to deoxythymidine monophosphate
(dTMP), which is then phosphorylated to deoxythymidine triphosphate
(dTTP). dTTP is required for DNA synthesis; therefore, in vitamin B-12
deficiency, formation of dTTP and accumulation of 5-methyl-THF is
inadequate, trapping folate in its unusable form and leading to retarded
DNA synthesis. RNA contains dUTP (deoxyuracil triphosphate) instead of
dTTP, allowing for protein synthesis to proceed uninterrupted and
resulting in macrocytosis and cytonuclear dissociation.
Because folate deficiency causes macrocytosis and cytonuclear
dissociation via the same mechanisms, both deficiencies lead to
megaloblastic anemia and disordered maturation in granulocytic lineages;
therefore, folate supplementation can reverse the hematologic
abnormalities of vitamin B-12 deficiency but has no impact on the
neurologic abnormalities of vitamin B-12 deficiency, indicating both
result from different mechanisms.
Vitamin B-12 role in the peripheral and central nervous systems
The neurologic manifestation of
cobalamin deficiency
is less well understood. CNS demyelination may play a role, but how
cobalamin deficiency leads to demyelination remains unclear. Reduced SAM
or elevated methylmalonic acid (MMA) may be involved.
SAM is required as the methyl donor in polyamine synthesis and
transmethylation reactions. Methylation reactions are needed for myelin
maintenance and synthesis. SAM deficiency results in abnormal
methylated phospholipids such as phosphatidylcholine, and it is linked
to central myelin defects and abnormal neuronal conduction, which may
account for the encephalopathy and myelopathy. In addition, SAM
influences serotonin, norepinephrine, and dopamine synthesis. This
suggests that, in addition to structural consequences of vitamin B-12
deficiency, functional effects on neurotransmitter synthesis that may be
relevant to mental status changes may occur. Parenthetically, SAM is
being studied as a potential antidepressant.
Another possible cause of neurologic manifestations involves
the other metabolically active form of cobalamin, adenosylcobalamin (see
image below), a mitochondrial cofactor in the conversion of
L-methylmalonyl CoA to succinyl CoA. Vitamin B-12 deficiency leads to an
increase in L-methylmalonyl-CoA, which is converted to D-methylmalonyl
CoA and hydrolyzed to MMA. Elevated MMA results in abnormal odd chain
and branched chain fatty acids with subsequent abnormal myelination,
possibly leading to defective nerve transmission.
 Vitamin B-12–associated neurological
diseases. Cobalamin deficiency leads to reduced adenosylcobalamin, which
is required for production of succinyl-CoA. D-methylmalonyl-CoA is
converted to methylmalonic acid.
Vitamin B-12–associated neurological
diseases. Cobalamin deficiency leads to reduced adenosylcobalamin, which
is required for production of succinyl-CoA. D-methylmalonyl-CoA is
converted to methylmalonic acid.
More recent studies propose a very different paradigm: B-12
and its deficiency impact a network of cytokines and growth factors, ie,
brain, spinal cord, and CSF TNF-alpha; nerve growth factor (NGF), IL-6
and epidermal growth factor (EGF), some of which are neurotrophic,
others neurotoxic. Vitamin B-12 regulates IL-6 levels in rodent CSF. In
rodent models of B-12 deficiency parenteral EGF or anti-NGF antibody
injection prevents, like B-12 itself, the SCD-like lesions.
In the same models, the mRNAs of several cell-type specific
proteins (glial fibrillary acidic protein, myelin basic protein) are
decreased in a region specific manner in the CNS, but, in the PNS
myelin, protein zero and peripheral myelin protein 22 mRNA remain
unaltered.
In human and rodent serum and CSF, concomitantly with a
vitamin B-12 decrease, EGF levels are decreased, while at the same time,
TNF-alpha increases in step with homocysteine levels. These
observations provide evidence that the clinical and histological changes
of vitamin B-12 deficiency may result from up-regulation of neurotoxic
cytokines and down-regulation of neurotrophic factors.
[12]
N
N
2 O can oxidize the cobalt core of vitamin B-12 from a 1
+ to 3
+
valance state, rendering methylcobalamin inactive, inhibiting HC
conversion to methionine and depleting the supply of SAM. Patients with
sufficient vitamin B-12 body stores can maintain cellular functions
after N
2 O exposure, but in patients with borderline or low
vitamin B-12 stores, this oxidation may be sufficient to precipitate
clinical manifestations.
Epidemiology
Frequency
The prevalence of vitamin B-12 deficiency is difficult to
ascertain because of diverse etiologies and different assays (i.e.,
radioassay or chemiluminescence). Affected individuals may number
300,000 to 3 million in the United States.
Using the radioassay and a value less than 200 pg/mL, the
prevalence of vitamin B-12 deficiency is 3-16%. In a geriatric
population using a radioassay cutoff of 300 pg/mL and elevated HC and
MMA levels, a prevalence of 21% was reported.
Of HIV-seropositive individuals, 11% are vitamin B-12
deficient; another 12% have levels of 200-240 pg/mL. In a subgroup with
chronic diarrhea, the rate reaches 39%. However, the importance for
vitamin B-12 deficiency in the development of neurologic disease in
these patients remains unclear.
In Europe, the prevalence of vitamin B-12 deficiency is 1.6-10%.
In India, a hospital population radioassay study with a cutoff
of 200 pg/mL found a vitamin B-12 deficiency in 0.88% of patients, with
borderline values in 3.8%.
Mortality/Morbidity
See the list below:
-
Vitamin B-12 deficiency is associated with an elevated HC.
-
The prevalence of hyperhomocysteinemia in the general
population is 5-10%; in people older than 65 years it may reach 30-40%.
Elevated HC is a risk factor for coronary artery, cerebrovascular, and
peripheral vascular diseases and venous thrombosis. About 10% of the
vascular disease risk in the general population is linked to HC.
-
Case-control studies have reported a correlation between
multi-infarct dementia or dementia of the Alzheimer type and elevated
HC; vitamin B-12 supplementation had no clinical benefit.
-
Neural tube defects are associated with low folate and vitamin B-12.
-
PA patients have a 3 times and 13 times increased risk of
gastric carcinoma and gastric carcinoid tumors, respectively.
-
Patients with diabetes mellitus type 1 and autoimmune thyroid
diseases are at higher risk of developing PA. A 2009 study noted a 22%
prevalence of vitamin B-12 deficiency in patients with diabetes mellitus
type 2.
[13]
-
Multifactorial abnormalities of vitamin B-12 metabolism and absorption occur in HIV infection.
Race
See the list below:
-
PA prevalence may be higher in white people and lower in Hispanic and black people.
-
No known relationship exists between neurologic symptoms and race.
-
Studies in Africa and the United States have shown higher
vitamin B-12 and transcobalamin II levels in black than in white
individuals. Additionally, blacks have lower HC levels and metabolize it
more efficiently than whites.
Sex
See the list below:
-
In Europe and Africa, the prevalence of PA is higher in
elderly women than men (1.5:1), while in the United States no
differences exist.
-
Men have higher HC levels at all ages.
-
Pregnancy and estrogen replacement in postmenopausal women lower HC levels.
Age
See the list below:
-
PA occurs in people of all ages, but it is more common in
people older than 40-70 years and, in particular, in people older than
65 years.
-
In white people, the mean age of onset is 60; in black people, the mean age is 50 years.
-
Congenital PA manifests in children aged 9 months to 10 years; the mean age is 2 years.
History
Clinical course
The neurologic features are attributable to pathology in the
peripheral and optic nerves, posterior and lateral columns of the spinal
cord (subacute combined degeneration), and in the brain. Interestingly,
hematologic and neurologic manifestations are occasionally dissociated.
[14, 15] An
inverse correlation in the severity of both manifestations has been
suggested. In patients with neuropsychiatric abnormalities, 28% lack
anemia or macrocytosis.
Clinical manifestations due to vitamin B-12 deficiency are
unrelated to etiology. In a prospective comparative study between
antiparietal cell antibody positive and negative patients, no
significant difference was shown in clinical, electrodiagnostic, and
radiological features.
[16]
Although the clinical features of vitamin B-12 deficiency may
consist of a classic triad of weakness, sore tongue, and paresthesias,
these are not usually the chief symptoms.
Onset is subacute or gradual, although more acute courses have been described, in particular after N
2 O
exposure. In 1986, Schilling described 2 patients with unrecognized
vitamin B-12 deficiency who developed paresthesias and poor manual
dexterity 1-3 months after brief N
2 O exposure.
[17] In 1995, Kinsella and Green described a 70-year-old man with paresthesias and hand clumsiness after 2 exposures to N
2 O over 3 months.
[18]
Onset is often with a sensation of cold, numbness, or
tightness in the tips of the toes and then in the fingertips, rarely
with lancinating pains. Simultaneous involvement of arms and legs is
uncommon, and onset in the arms is even rarer.
Paresthesias are ascending and occasionally involve the trunk, leading to a sensation of constriction in the abdomen and chest.
Untreated patients may develop limb weakness and ataxia.
In 1991, Healton et al performed detailed neurologic
evaluations of 143 patients with vitamin B-12 deficiency; 74% presented
with neurologic symptoms.
[19]
-
Isolated numbness or paresthesias were present in 33%.
-
Gait abnormalities occurred in 12%.
-
Psychiatric or cognitive symptoms were noted in 3%.
-
Visual symptoms were reported in 0.5%. Symptoms include
subacute progressive decrease in visual acuity, usually caused by
bilateral optic neuropathy and rarely pseudotumor cerebri or optic
neuritis.
-
Rare autonomic features include orthostasis, sexual dysfunction, and bowel and bladder incontinence.
-
Other symptoms include lightheadedness and impaired taste and smell.
-
Asymptomatic neurologic manifestations can be detected using somatosensory evoked potentials (SSEP); see below.
-
Nonneurologic symptoms, some of which may also reflect autonomic nervous system involvement, were present in 26%.
- Constitutional symptoms, including anorexia and weight
loss occurred in 50%. Low-grade fever that resolves with treatment
occurred in 33% of cases. Other symptoms include fatigue and malaise.
- Cardiovascular symptoms include syncope, dyspnea, orthopnea, palpitations, and angina.
- Gastrointestinal symptoms include heartburn, flatulence, constipation, diarrhea, sore tongue, and early satiety.
In a prospective study of 57 patients with vitamin B-12
deficiency neurological syndrome, common presenting syndromes included
myeloneuropathy (25), myelopathy (14), myeloneuroencephalopathy (13),
myeloencephalopathy (4), and behavioral (1).
[126]
Physical
Most patients exhibit signs of peripheral nervous system (PNS)
or spinal cord involvement, but the extent of PNS involvement remains
unclear, in part because both neuropathy and myelopathy can cause
impaired vibration sense, ataxia, and paresthesias. Either can be
affected first in the early stages. Objective sensory abnormalities
usually result from posterior column involvement and less often from PNS
disease.
In 1919, Woltmann found features of PNS disease in 4.9% of
patients with PA, including distal hyporeflexia or areflexia; 80% of
these also had evidence of cord involvement.
[20]
In 1991, Healton summarized his experience with a large group of patients as follows:
[19]
-
Isolated neuropathy was reported in 25% of patients.
-
Myelopathy occurred in 12% of cases.
-
A combination of neuropathy and myelopathy was noted in 41%.
-
Neuropsychiatric manifestations, such as recent memory loss
with reduced attention span and otherwise normal cognition, depression,
hypomania, paranoid psychosis with auditory or visual hallucinations
(megaloblastic madness), violent behavior, personality changes, blunted
affect, and emotional liability, were reported in 8% of patients.
-
Ocular findings included a cecocentral scotoma and occurred in
0.5% of cases. Others have described optic atrophy, nystagmus, small
reactive pupils, and chiasmatic lesion causing bitemporal hemianopia.
-
Normal findings were noted on neurologic examination in 14% of patients despite paresthetic symptoms.
Early in the course, poor joint position and vibration sense
predominate. Typically, the legs are affected before the arms. Rarely
are all limbs affected simultaneously. A Romberg sign is commonly found.
The gait may be wide based.
On presentation, 50% of patients have absent ankle reflexes
with relative hyperreflexia at the knees. Plantars are initially flexor
and later extensor. A Hoffman sign may be found.
As the disease progresses, ascending loss of pinprick, light
touch, and temperature sensation occurs. Later, depending on the
predominance of posterior column versus cortical spinal tract
involvement, ataxia or spastic paraplegia predominates. Then, PNS
involvement causes distal limb atrophy.
Cognitive testing may reveal mild impairment or frank dementia.
Nonneurologic manifestations include the following:
-
General - Lemon-yellow waxy pallor, premature whitening of
hair, flabby bulky frame, mild icterus, and blotchy skin pigmentation in
dark-skinned patients
-
Cardiovascular - Tachycardia, congestive heart failure
-
Gastrointestinal - Beefy, red, smooth, and sore tongue with loss of papillae that is more pronounced along edges
Abnormal vitamin B-12 metabolism occurs in infants born to
vitamin B-12–deficient mothers or those with hereditary diseases,
including the Imerslünd-Grasbeck syndrome (cublin mutation resulting in
decreased cobalamin transport from the intestinal lumen), transcobalamin
II deficiency, and intracellular cobalamin abnormalities (classified as
Cbl A though G with neurologic features in Cbl C and Cbl D, see below).
Symptoms become prominent after exhaustion of vitamin B-12 stores
acquired in utero. Infants present with developmental delay, failure to
thrive, lethargy, poor feeding, mental retardation, seizures,
listlessness, irritability, ataxia, hyporeflexia, hypotonia, pathologic
reflexes, coma, tremor, and myoclonus. The latter may worsen transiently
upon initiation of treatment.
Causes
Inadequate vitamin B-12 absorption is the major pathomechanism and may result from several factors.
-
Intrinsic factor deficiency
- PA accounts for 75% of cases of vitamin B-12 deficiency.
It is an autoimmune attack on gastric IF. Antibodies are present in 70%
of patients. They may block the formation of the cobalamin-IF complex or
block its binding with cublin. Other antibodies are directed at
parietal cell hydrogen-potassium adenosine triphosphatase (ATPase).
- Juvenile PA results from inability to secrete IF.
Secretion of hydrogen ions and the gastric mucosa are normal.
Transmittance is autosomal recessive inheritance of abnormal GIF on
chromosome arm 11q13.
- Destruction of gastric mucosa can occur from gastrectomy or Helicobacter pylori infection. A Turkish study found endoscopic evidence of H pylori infection in more than 50% of vitamin B-12–deficient patients. Antibiotics alone eradicated H pylori in 31 patients, with resolution of vitamin B-12 deficiency.
-
Deficient vitamin B-12 intake: Intake may be inadequate
because of strict vegetarianism (rare), breastfeeding of infants by
vegan mothers, alcoholism, or following dietary fads.
-
Disorders of terminal ileum: Tropical sprue, celiac disease,
enteritis, exudative enteropathy, intestinal resection, Whipple disease,
ileal tuberculosis, and cublin gene mutation on chromosome arm 10p12.1
in the region designated MGA 1, which affects binding of the
cobalamin-IF complex to intestinal mucosa (Imerslünd-Grasbeck syndrome),
are disorders that affect the terminal ileum.
-
Competition for cobalamin: Competition for cobalamin may occur in blind loop syndrome or with fish tapeworm (
Diphyllobothrium latum).
-
Abnormalities related to protein digestion related to
achlorhydria: Abnormalities include atrophic gastritis, pancreatic
deficiency, proton pump inhibitor use, and Zollinger-Ellison syndrome,
in which the acidic pH of the distal small intestine does not allow the
cobalamin-IF complex to bind with cublin.
-
Medications: Medications include colchicine, neomycin, and
p -aminosalicylic acid.
-
Transport protein abnormality: Abnormalities include
transcobalamin II deficiency (autosomal recessive inheritance of an
abnormal
TCN2 gene on chromosome arm 22q11.2-qter resulting in
failure to absorb and transport cobalamin) and deficiency of R-binder
cobalamin enzyme.
-
Disorders of intracellular cobalamin metabolism: These
disorders result in methylmalonic aciduria and homocystinuria in
infants.
- Isolated methylmalonic aciduria
- Cbl A is due to deficiency of mitochondrial cobalamin reductase resulting in deficiency of adenosylcobalamin.
- Cbl B is due to deficiency of adenosylcobalamin transferase resulting in deficiency of adenosylcobalamin.
- Methylmalonic aciduria and homocystinuria
- Cbl C is a combined deficiency of methylmalonyl CoA
mutase and homocysteine:methyltetrahydrofolate methyltransferase.
Patients have prominent neurologic features and megaloblastic anemia.
- Cbl D is a deficiency of cobalamin reductase. Patients have prominent neurologic features.
- Cbl F is a defect in lysosomal release of cobalamin.
- Isolated homocystinuria
- Cbl E is due to a defect in methionine synthase reductase located on chromosome arm 5p15.3-p15.2.
- Cbl G is due to a defect in methyltetrahydrofolate homocysteine methyltransferase located on chromosome arm 1q43.
-
Increased vitamin B-12 requirement: Requirement is increased in hyperthyroidism and alpha thalassemia.
-
Other causes
- In AIDS, vitamin B-12 deficiency is not infrequent.
Although the exact etiology remains obscure, it is likely a multimodal
process involving poor nutrition, chronic diarrhea, ileal dysfunction,
and exudative enteropathy. Low vitamin B-12 levels may be more common in
late than in early HIV disease.
- N 2 O exposure can occur iatrogenically (ie, anesthesia) or through abuse ("whippets").
Differential Diagnoses
Laboratory Studies
See the list below:
Clinical evidence of vitamin B-12 deficiency
- Serum cobalamin levels are the initial test.
- Two
assays exist: radioassay and the nonradioisotopic assay,
chemiluminescence, which is becoming more popular because of improved
automation, safety, and cost. Chemiluminescence has a higher reference
range value, from 250-1100 pg/mL versus 170-900 pg/mL for radioassay.
Using the radioassay and elevated homocysteine (HC) and methylmalonic
acid (MMA) as criterion standards, levels are less than 200 pg/mL in
90-95% of patients, 200-300 pg/mL in 5-10%, and greater than 300 pg/mL
in 0.1-1%. Be aware of the assay used and how the reference range was
determined. A serum cobalamin level that is within the reference range
does not exclude cobalamin deficiency.
Abnormally low vitamin B-12 levels: Test for PA by measuring antibodies against IF.
- Antiparietal
cell antibodies are present in 90% of PA cases. In patients older than
70 years, 10% have false-positive abnormal antibody levels.
- IF antibodies are present in 60% of patients. These are more specific but less sensitive.
- If either antibody is positive, the diagnosis of PA is confirmed and further testing is not required.
- If
antibodies are negative, obtain a serum gastrin level to test for
achlorhydria, which is associated with PA. If these are elevated, the
diagnosis is likely PA. If these results are normal, perform a Schilling
test.
Borderline
vitamin B-12 level and clinical features of vitamin B-12 deficiency:
Measure methylmalonic acid (MMA) and homocysteine (HC).
- Both folate and vitamin B-12 deficiency can lead to metabolite elevation.
- In vitamin B-12 deficiency, MMA and HC are elevated, although HC elevation occurs by itself. MMA is more sensitive than HC.
- In folate deficiency, MMA is within the reference range and HC is elevated.
- MMA
and HC are considered abnormal when greater than 3 standard deviations
above the mean. Reference range values are not age dependent for MMA and
are 70-350 nM/L. For patients younger than 60 years, reference range
values are 5-15 µM/L for HC. In people older than 60 years, the cutoff
for HC is 20 µM/L.
- If both metabolites are within the reference
range, vitamin B-12 deficiency is effectively ruled out. Only 0.2% of
400 patients with low serum vitamin B-12 had normal metabolite levels,
and 10% of 98 patients with folate deficiency had metabolite levels
within the reference range. False-positive elevations in MMA and HC
occur in inborn errors of metabolism, renal disease, and deficiencies of
folate. If either metabolite is elevated, test for PA or use the
Schilling test.
Schilling
test: The Schilling test is used to determine the etiology of vitamin
B-12 deficiency in patients with normal IF antibodies.
- Stage 1:
Administer radiolabeled cobalamin 0.5-2.0 mCi PO to fasted patients. One
to 6 hours later, administer unlabeled cobalamin 1000 mcg IM to
saturate transcobalamin and flush hepatic storage. Measure the
percentage of radiolabeled cobalamin in a 24-hour urine specimen.
Urinary excretion within the reference range is 10-35% over 24 hours.
Reduced urinary excretion of cobalamin, ie, less than 7-9% based on
individual laboratory reference range values, in persons with normal
renal function supports decreased absorption of oral cobalamin. If
excretion is low, proceed to stage 2.
- Stage 2: Stage 1 is
repeated with coadministration of porcine IF 60 mg. If the absorption of
cobalamin is normalized, the presumptive diagnosis is PA. If poor
absorption persists after administration, proceed to stage 3.
- Stage 3: Tetracycline is administered for 5 days prior to reperformance of stage 1 to exclude blind loop as the etiology.
- Stage 4: Pancreatic enzymes are administered with repetition of stage 1 to test for pancreatic disease.
- Caveats:
If vitamin B-12 is administered 48 hours before the Schilling test,
dilution of the radiolabeled cobalamin and spuriously low apparent
urinary excretion and false-positive results occur. False-negative
values occur in food-bound malabsorption due to achlorhydria. True
negative results are from dietary deficiencies (vegan) and cobalamin
binding–protein abnormalities.
Routine hematologic and chemistry tests
- Hematologic abnormalities may be absent at the time of neurologic presentation.
- Vitamin
B-12 deficiency produces the classic picture of macrocytic anemia, with
a mean corpuscular value (MCV) greater than 100 fL. The MCV correlates
with estimated vitamin B-12 level:
- MCV of 80-100 fL (normal) indicates less than 25% probability of vitamin B-12 deficiency.
- MCV of 115-129 fL indicates a 50% probability.
- MCV greater than 130 fL indicates a 100% probability.
- Peripheral
blood smear shows macro-ovalocytosis, anisocytosis, and poikilocytosis,
as well as basophilic stippling of the erythrocytes and Howell-Jolly
bodies. Reticulocyte count can be within the reference range or low.
Hypersegmentation (>5% of neutrophils with > 5 lobes or 1% with
> 6 lobes) of polymorphonuclear cells may occur without anemia.
Thrombocytopenia is observed in approximately 50% of patients, and
platelets often have bizarre size and shape.
- Serum indirect bilirubin and lactate dehydrogenase (LDH) may be elevated because PA can have a hemolytic component.
- Achlorhydria is present in many patients with PA.
Laboratory parameters after administration of vitamin B-12
- Anemic patients
- Reticulocytosis starts in 3-4 days and peaks at 1 week.
- Hemoglobin concentration rises in 10 days and returns to the reference range in 8 weeks.
- LDH falls within 2 days.
- Hypersegmented neutrophils disappear in 1-2 weeks.
- Patients with severe anemia and borderline-to-low iron stores
- Serum iron level falls within 24 hours because of increased erythropoiesis.
- Hypokalemia may develop because of increased potassium utilization in hematopoiesis.
Imaging Studies
See the list below:
Because of the increased incidence of gastric cancer in PA, gastric radiographic series are suggested at the first visit.
In
patients with myelopathy, MRI may reveal regional T2 and
fluid-attenuated inversion recovery (FLAIR) hyperintensities mainly in
the thoracic posterior columns with possible extension into the brain
stem. In patients with chronic disease, atrophy of the spinal cord is
observed.
Brain MRI may show T2 and FLAIR hyperintensities in the cerebral white matter and around the fourth ventricle.
Brain MRI of infants with vitamin B-12 deficiency may show delayed myelination.
Other Tests
See the list below:
Abnormal
evoked potentials may be the first electrodiagnostic finding, even in
asymptomatic patients with normal neurologic examination findings. The
abnormalities are often referable to a central conduction defect;
however, peripheral nerves are also affected.
Somatosensory
evoked potentials (SSEP) may reveal prolongation of L3-P27 latency,
reflecting a defect in conduction in the large-fiber sensory pathway
between the cauda equina and the contralateral sensory cortex.
Visual evoked potential (VEP) findings are as follows:
- VEP findings may be abnormal even without visual symptoms or signs.
- Prolongation of the P100 waveform can be unilateral or bilateral.
- P100 may normalize after cobalamin replacement.
Nerve conduction study (NCS)/EMG findings are as follows:
- In 1943, Dynes and Norcross found evidence of neuropathy in 23% of patients with PA.[21]
- In a recent study, up to 65% of untreated patients had peripheral neuropathy.
- Axonal sensorimotor polyneuropathy is present in up to 80%. Demyelinating or mixed forms are less frequent.
- Typical features are decreased conduction velocities and motor or sensory amplitude and denervation on EMG.
- In
1991, Healton et al found decreased motor nerve conduction velocities
(NCV), absent or reduced sensory potentials, and fibrillations in the
distal muscles indicating mixed demyelinating-axonal disease in 7 of 9
cases of vitamin B-12 deficiency and neuropathy.[19]
- In 1998, Steiner et al described 5 patients with demyelinating polyneuropathy.[22]
- Sensory
nerves are usually more affected than motor nerves and are more
severely affected distally than proximally. Proximal focal conduction
block has been reported, which reversed on treatment.
Electroencephalography
(EEG) findings may be normal or show nonspecific slowing. Follow-up EEG
findings may be improved in response to treatment.
Histologic Findings
The
CNS is better characterized than the PNS in vitamin B-12 deficiency.
The classic picture is subacute combined degeneration of the spinal cord
involving the dorsal columns and corticospinal tracts. Lesions are
concentrated in the cervical and upper thoracic cord and the cerebrum.
Spinal cord findings
See the list below:
Brain findings
See the list below:
Macroscopic: The brain appears normal.
Microscopic:
Findings resemble the spinal cord pathology with scanty small
perivascular foci of demyelination within the white matter featuring
myelin swelling and axon degeneration. The optic nerve typically shows
degeneration, predominantly in the papillomacular bundles.
Peripheral nervous system findings
See the list below:
Unlike
EMG/NCS findings, which indicate predominantly axonal or mixed
axonal-demyelinating neuropathy, histologic examination suggests either
primary demyelinating or axonal neuropathy with secondary demyelination.
However, most studies were performed before the introduction of
semithin, teased fiber, and electron microscopy analysis.
- A biopsy of the anterior tibial nerve recorded loss of myelin sheath and no loss of axons.[23]
- A sural biopsy revealed loss of myelinated fibers and no demyelination in teased fibers, indicating secondary demyelination.[24]
- In
1959, Coers and Woolf found endplate and preterminal axon abnormalities
in muscle biopsies of patients with cobalamin deficiency, suggesting
axonal damage.[25]
The
pathology is better described in animal models. In cobalamin-deficient
rats, electron microscopy revealed intramyelin and endoneural edema,
with no or minimal axonal damage (reversible on administration of
cobalamin) in nerves, dorsal root ganglia, and the ventral and dorsal
roots.
Dalla Torre et al report 2
patients with isolated sensory axonal neuropathy secondary to vitamin
B12 deficiency who recovered after cyanocobalamin replacement.
[26]
Nonneurologic findings
See the list below:
Bone marrow hyperplasia
Mild-to-moderate splenomegaly
Increased iron deposits in the reticuloendothelial system
Atrophy in all layers of the stomach, sparing the pyloroduodenal region
Medical Care
See the list below:
Establish the diagnosis and etiology of vitamin B-12 deficiency and treat with adequate doses.
[27]
The
consequences of vitamin B-12 deficiency, encephalopathy, myelopathy,
and peripheral and optic neuropathy require adequate medical care.
Physical
therapy and occupational therapy are needed to improve gait, balance,
and arm function. Patients may require canes or a walker for ambulation
and safety.
In patients with
encephalopathy, neuropsychological interventions may improve cognition,
social functioning, and interpersonal relationships.
Patients
with PA are at increased risk for gastric carcinoma, colorectal
adenocarcinoma, and carcinoid tumors and must be monitored.
Consultations
Consultations with a gastroenterologist, a hematologist, and a neurologist must be considered.
Diet
When
the cause of vitamin B-12 deficiency is low intake, recommend that
patients eat food that contains vitamin B-12 such as meat, eggs, cheese,
and yogurt. Supplementation is required when religious or cultural
restrictions render dietary changes impossible.
Activity
In
most patients with vitamin B-12–associated neuropathy/myelopathy, no
restriction on physical activity is necessary unless weakness or gait
ataxia is severe. Also, severe encephalopathy may lead to 24-hour
supervision. In severe anemia or congestive heart failure, the patient
should limit strenuous exercise.
Medication Summary
Standard treatment in patients with vitamin B-12 deficiency
consists of parenteral or oral cobalamin. The hematologic abnormalities
may respond to folate, but the neurologic manifestations only respond to
cobalamin.
Numerous treatment regimens have been proposed, including
cobalamin 1000 mcg IM/SC daily for 5 days followed by 1000 mcg/wk for 5
weeks, then 100-1000 mcg/mo for life.
Because 1% of cobalamin is absorbed by passive diffusion,
administration of large oral doses is an alternative; 1000 mcg daily
yields a daily absorption of 10 mcg, which exceeds the 2-mcg recommended
daily allowance (RDA) requirement.
In addition to cobalamin replacement, oral IF supplementation
is being evaluated. Supplementation with SAM or methionine-rich diets
are being studied for N 2 O-induced myeloneuropathies.
Diagnosis and treatment of tapeworm infection and celiac and
Crohn diseases can improve intestinal vitamin B-12 malabsorption. With
blind loop syndrome, tetracycline can normalize the intestinal flora and
vitamin B-12 absorption.
Dietary supplements
Class Summary
Cyanocobalamin is used to replenish the deficiency caused by any of the etiologies described.
Most stable and available form of vitamin B-12. Absorbed rapidly to the organism from IM or SC applications.
Oral cyanocobalamin can replace parenteral formulations. Is
effective in PA because 1% of free cobalamin is absorbed via diffusion
rather than requiring the presence of IF.
Folate supplementation can reverse the hematologic abnormalities, but the neurologic manifestations only respond to cobalamin.
Further Outpatient Care
Patients
with neurologic impairment may require additional care in skilled
nursing units or rehabilitation facilities. Outpatient follow-up is
required to ensure response to therapy.
Further Inpatient Care
Once
therapy is initiated, hospitalization is only required for patients
with life-threatening anemia or with severe neurologic deficits
requiring supervision or rehabilitation.
Inpatient & Outpatient Medications
See the list below:
Cobalamin 100-1000 mcg/mo SC/IM, 1000 mcg/d PO is provided for lifelong maintenance.
Compliance must be verified to avoid recurrence of symptoms.
Deterrence/Prevention
See the list below:
Relatives of patients with PA must be made aware of the increased familial incidence.
Individuals
with total gastrectomy, pancreatectomy, or atrophic gastritis should
undergo periodic testing for vitamin B-12 deficiency.
Testing
vitamin B-12 (and folate) levels in elderly patients is good practice
because of the high incidence of deficiencies. Asymptomatic deficiency
should be worked up and treated.
Strict vegetarians should supplement vitamin B-12 in their diets.
Complications
See the list below:
If left untreated, neurologic complications worsen.
Severe anemia may lead to congestive heart failure.
Incidence of atrophic gastritis, gastric carcinoma, and carcinoid tumors is increased in patients with PA.
Patients
with PA are at increased risk for other autoimmune disorders, such as
myasthenia gravis, Lambert-Eaton myasthenic syndrome, type 1 diabetes
mellitus, Hashimoto thyroiditis, hypogammaglobulinemia, vitiligo, and
rheumatoid arthritis.
[28]
Risk of neural tube defects is increased in untreated pregnant women.
Prognosis
See the list below:
Therapy with vitamin B-12 in subacute combined degeneration stops progression and improves neurologic deficit in most patients.
Younger patients with less severe disease and short duration illness do better.
In
a large retrospective review of 57 patients with subacute combined
degeneration, absence of sensory level, absent Rhomberg sign, and flexor
planter reflex were associated with good prognosis.
[29]
On
spinal MRI, involvement of less than 7 spinal segments, cord swelling,
and enhancement, but not cord atrophy, were associated with better
prognosis.
[29]
Clinical improvement is most pronounced in the first 2 months but continues up to 6 months.